
Abstract
Introduction
The objective of this study was to evaluate the cost-effectiveness of lisocabtagene maraleucel (liso-cel) versus other available chimeric antigen receptor T-cell therapies, including axicabtagene ciloleucel (axi-cel) and tisagenlecleucel (tisa-cel), in patients who had received at least two prior therapies from a United States (US) commercial third-party payer perspective.
Methods
To capture this heterogeneity in survival outcomes, we used mixture cure models to extrapolate progression-free survival (PFS) and overall survival (OS). Patient-level data from TRANSCEND NHL 001 for liso-cel and reconstructed patient-level data from ZUMA-1 for axi-cel, JULIET for tisa-cel, and SCHOLAR-1 for salvage chemotherapy, derived using the Guyot method, were used for OS and PFS. The model included adverse events associated with liso-cel, axi-cel, and tisa-cel.
Results
Liso-cel was less costly (incremental cost of − $74,980) and marginally more effective (0.002 incremental quality-adjusted life-years [QALY]) than axi-cel and had an incremental cost of $67,925 and 2.02 incremental QALYs over tisa-cel in the base case. Results remained consistent in sensitivity analyses, with the liso-cel OS cure fraction being the main driver of cost-effectiveness compared with both axi-cel and tisa-cel.
Conclusion
This analysis estimated that liso-cel is cost-effective compared with tisa-cel and axi-cel from a commercial US payer perspective.
Similar content being viewed by others

Why carry out this study? |
The comparative cost-effectiveness of the marketed chimeric antigen receptor (CAR) T-cell therapies lisocabtagene maraleucel (liso-cel), axicabtagene ciloleucel (axi-cel), and tisagenlecleucel (tisa-cel) in patients with large B-cell lymphoma who had received at least two prior therapies from a US commercial third-party payer perspective was unknown. |
What was learned from the study? |
Liso-cel is estimated to be cost-effective compared with the marketed CAR T-cell therapies axi-cel and tisa-cel in this patient population and setting. |
Compared with axi-cel, liso-cel generated similar quality-adjusted life-years at lower cost, partly owing to lower rates of highly burdensome adverse events of special interest for CAR T-cell therapies. |
Compared with tisa-cel, liso-cel generated greater quality-adjusted life-years owing to superior survival, but at slightly higher cost because of higher drug acquisition cost. |
Introduction
Large B-cell lymphoma (LBCL) is the most common, aggressive subtype of non-Hodgkin lymphoma. Although a majority of patients can achieve long-term remission after first-line therapy for LBCL [1], approximately 40% exhibit relapsed or refractory (R/R) disease [2,3,4]. Salvage chemoimmunotherapy combination regimens, with or without autologous stem cell transplantation, have conventionally been the standard of care for R/R LBCL; however, patients who progress after at least two lines of treatment are unlikely to benefit from additional chemoimmunotherapy [5], with poor response rates observed with salvage chemotherapy in third-line or later (3L+) LBCL [1].
Chimeric antigen receptor (CAR) T-cell therapies, generated from genetically engineered autologous T-cells to express anti-CD19 CARs, revolutionized the treatment landscape for 3L+ LBCL with United States (US) Food and Drug Administration approval of axicabtagene ciloleucel (axi-cel) [6], tisagenlecleucel (tisa-cel) [7], and lisocabtagene maraleucel (liso-cel) [8].
CAR T-cell therapies have shown durable responses and improved survival outcomes compared with historical cohorts treated with conventional chemotherapy in patients with 3L+ LBCL [9,10,11] and offer potential long-term remission for these patients; however, these therapies have high costs and logistic complexities for the treatment center, and clinical practitioners must identify patients for whom this therapy is suitable and for whom the therapeutic benefits outweigh potential risks [12]. It is important for clinical and payer decision-makers to understand the value of the different CAR T-cell therapy options. The study objective was to evaluate the cost-effectiveness of liso-cel versus axi-cel and tisa-cel in the 3L+ setting, from a US commercial third-party payer perspective.
Methods
Overview
In line with other published models of CAR T-cell therapies [13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28], a Microsoft Excel®-based partitioned survival model was developed to estimate the cost-effectiveness of liso-cel versus axi-cel and tisa-cel in accordance with International Society for Pharmacoeconomics and Outcomes Research good modeling practice guidelines [29]. The modeled population was adults with R/R LBCL after at least two prior therapies including an anthracycline and rituximab (or other CD20-targeted agent), per the TRANSCEND NHL 001 (TRANSCEND) trial [30]. The model assumed the starting age was 60 years and 36% of patients were female, per TRANSCEND. The analysis considered direct medical costs only. A lifetime horizon (up to 50 years) was used to fully capture outcomes, and an annual discount rate of 3% was applied to cost and health outcomes as recommended by the Second Panel on Cost-Effectiveness in Health and Medicine [31]. The main outcome was incremental cost per quality-adjusted life-year (QALY) gained.
Kaplan-Meier survival curves for CAR T-cell therapies in 3L+ LBCL exhibit a plateau [32,33,34] in the long-term follow-up, indicating a fraction of patients may achieve durable remission. To capture this heterogeneity in survival outcomes, we used mixture cure models (MCM) to extrapolate progression-free survival (PFS) and overall survival (OS). MCMs assume that the population is a mixture of noncured (worse prognosis; shorter OS) and cured (better prognosis; OS similar to general population) patients. MCM simultaneously estimates the fraction of the studied population achieving cure and survival of those not achieving cure using parametric survival distributions [35]. Parametric distributions were selected on the basis of goodness of fit criteria and clinical plausibility of long-term projections and cure fractions (OS vs PFS) for each treatment [36].
Model Structure
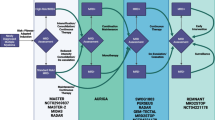
The model included three health states: PFS, progressed disease, and death (Fig. 1). OS projections were used to determine those alive (and dead) over time, and PFS projections were used to further partition patients into PFS and progressed disease states. Patients remaining progression-free beyond 2 years were assumed to require less-frequent monitoring over time, and patients who progressed were assumed to receive subsequent treatment. A weekly cycle length was used for the first 5 years to enable accurate calculation of costs and QALYs associated with the relatively high rate of events in this interval, after which an annual cycle length was used to simplify model calculations without jeopardizing accuracy.

Model structure. Patients intended to receive CAR T-cell therapy have a pretreatment period from leukapheresis until CAR T-cell infusion, during which patients receive lymphodepleting chemotherapy and bridging therapy (if needed). Some patients may not receive their CAR T-cell infusion owing to manufacturing errors, disease progression, or death; therefore, the model stratified patients according to the proportions who did not receive CAR T-cell infusion, per the respective trials, to accurately calculate the associated costs and outcomes before entering the partitioned survival model. CAR chimeric antigen receptor
Patients’ treatment experience from leukapheresis to CAR T-cell infusion can vary (Fig. 1). Patients who died before CAR T-cell infusion accrued QALYs during the pretreatment period and the cost of leukapheresis. Those who failed to receive CAR T-cell therapy for other reasons accrued costs and outcomes associated with salvage chemotherapy, the historical 3L+ standard of care, based on data from SCHOLAR-1 [1]. Total costs and outcomes of each treatment arm were an average of costs and outcomes for these cohorts and the respective CAR T-cell therapy, weighted according to this pretreatment period stratification as observed in the trials. Bridging chemotherapy (systemic, radiation, or both) for disease control during CAR T-cell product manufacturing is needed for some patients during the pre-infusion period. Bridging therapy protocols differed across clinical trials.
Model Inputs
Survival Projections and Comparative Efficacy
For OS and PFS, patient-level data from TRANSCEND were used for liso-cel. For axi-cel, tisa-cel, and salvage chemotherapy, reconstructed patient-level data from ZUMA-1 [37, 38], JULIET [39], and SCHOLAR-1 [1] were used, respectively. For axi-cel, OS is based on a later data cut than those for PFS and safety as the latter were not published with the most recent ZUMA-1 follow-up [33].
For liso-cel, gamma and log-logistic distributions were chosen to extrapolate OS and PFS, respectively. These were fit independently and were used as the reference curve to which relative treatment effects were applied to project PFS and OS for axi-cel and tisa-cel (Fig. 2). Two-dimensional treatment effects (i.e., the effect on the estimated cure fraction and survival of noncured patients) were estimated by fitting joint MCM. In the base case, liso-cel data incorporated weights derived from pairwise unanchored matching-adjusted indirect comparisons (MAIC) versus tisa-cel [40] and axi-cel [41] (Supplemental Table 1) to minimize bias induced when comparing these single-arm studies. It should be noted that the MAIC versus axi-cel used in the base case did not match on use of bridging therapy because of its impact on the effective sample size [41], though this was an important difference between ZUMA-1 and TRANSCEND (bridging therapy was permitted in TRANSCEND but not in ZUMA-1). MAIC that matched on bridging therapy was used in the scenario analysis. A naïve comparison was also conducted in a scenario analysis.

Long-term OS (A) and PFS projections (B). axi-cel axicabtagene ciloleucel, KM Kaplan-Meier, liso-cel lisocabtagene maraleucel, OS overall survival, PFS progression-free survival, tisa-cel tisagenlecleucel
OS of cured patients was simulated as the age- and sex-adjusted US general population mortality [42]. An excess mortality risk was also applied to account for any secondary malignancies and long-term adverse effects of cancer-specific treatment. A standardized mortality ratio of 1.40 was applied for the first 2 years, followed by a standardized mortality ratio of 1.18 for the remainder of the patients’ lifetimes [43].
Adverse Events
The model included adverse events (AE) associated with liso-cel, axi-cel, and tisa-cel but not subsequent treatment or regimens administered as pretreatment given these would have negligible impacts on incremental outcomes. In the base case, odds ratios estimated from MAICs of safety data from the trials from updates of previously published analyses [40, 41] were applied to the liso-cel rates to derive AE rates for each comparator (Table 1). Observed rates for each comparator were used in scenario analysis.
The model included all grade 3 or higher AEs occurring in at least 5% of patients in any trial. Additionally, all-grade cytokine release syndrome (CRS), neurological events (NE), and hypogammaglobulinemia were included irrespective of incidence to capture resource use and health-related quality of life (HRQOL) associated with CAR T-cell therapy AEs of special interest (AESI) [30, 44]. Only grade 3 or higher AEs were assumed to impact HRQOL.
Utilities
Utility values were applied for the duration of the pretreatment period and the remaining time in each health state, with decrements due to AEs applied in PFS (Table 2). Utilities were estimated using EQ-5D data from TRANSCEND using a mixed-effects model for repeated measures (MMRM) that included baseline EQ-5D, AEs, and progressive disease as predictors. The analysis was conducted using US tariffs and a crosswalk algorithm [45] to convert from EQ-5D-5L to the EQ-5D-3L value sets.
Grade 3 or higher CRS disutility could not be estimated from TRANSCEND data owing to its low incidence and duration. Instead, a vignette-based time trade-off study designed to specifically estimate CRS disutility was used [46].
Resource Use and Costs
The model incorporated costs associated with CAR T-cell therapy pretreatment, treatment acquisition (using wholesale acquisition cost prices) and administration, postinfusion hospitalization, AE management, monitoring (by health state), subsequent treatment, and end-of-life costs. All costs were expressed as 2020 US dollars. Key cost inputs are presented in Table 2.
All patients accrued the cost of leukapheresis. In the base case, 62% of liso-cel–treated patients, 92% of tisa-cel–treated patients, and 0% of axi-cel–treated patients were assumed to receive bridging therapy per their respective trials, and these costs were reflected in the cost estimates. Lymphodepleting chemotherapy, modeled as one cycle of fludarabine plus an alkylating agent, was then applied for the patients who received CAR T-cell infusion [6, 7, 47].
Acquisition costs for liso-cel (50–110 × 106 CAR-positive T-cells), axi-cel (2 × 106 CAR-positive T-cells), tisa-cel (0.6–6.0 × 108 CAR-positive T-cells), and chemotherapies used for lymphodepletion, bridging, salvage (for those failing to receive CAR T-cell infusion), and subsequent treatment were sourced from the IBM® Micromedex® RED BOOK® pricing file [48]. Salvage chemotherapy was modeled as a mix of chemotherapy regimens (40% rituximab plus gemcitabine and oxaliplatin; 30% rituximab plus gemcitabine, dexamethasone, and cisplatin [R-GDP]; and 30% rituximab plus dexamethasone, cytarabine, and cisplatin [R-DHAP]). R-GDP and R-DHAP comprise the treatment arms in the LY.12 trial [49] (the largest contributing trial to SCHOLAR-1) [1].
Administration costs for CAR T-cell therapy were based on the infusion setting (inpatient vs outpatient) per the trials and prescribing information. For patients who received CAR T-cell therapy in the outpatient setting, cost estimation was based on analysis of Medicare 2019 claims data inflated to 2020. For patients who received infusions in the inpatient setting, the duration of the inpatient stay for CAR T-cell administration was assumed to be the same across all CAR T-cell therapies (11 days total) based on TRANSCEND, as differences in inpatient stays due to AEs were captured separately through treatment-specific AE rates. The cost per inpatient infusion was assumed to be the same as an outpatient administration plus the cost of one additional hospital bed day per the Healthcare Cost Utilization Project (HCUP) [50]. The cost of the remaining 10 days of the inpatient stay were based on the per-day cost from HCUP (Table 2). Patients in TRANSCEND who received nonconforming product accrued pretreatment, administration, and AE costs but did not accrue liso-cel acquisition costs.
Subsequent treatment was modeled as a mix of allogeneic hematopoietic stem cell transplantation, salvage chemotherapy, radiotherapy, and no active treatment, per TRANSCEND. This was assumed to be the same for all CAR T-cell therapies owing to the absence of published data from ZUMA-1 and JULIET. The distribution of salvage chemotherapy regimens was assumed to be the same as in the preprogression state.
For all-grade CRS and NEs and grade 3 or higher hypogammaglobulinemia, a microcosting approach was used. This considered the costs of drug therapy, diagnostics, and inpatient stays associated with the events. A similar approach was taken in other cost-effectiveness studies of CAR T-cell therapy [51, 52] because traditional costing approaches tend to underestimate the resources required to manage these AEs [53]. Microcosting inputs for CRS and NE were based on an analysis of the resources used for managing these events during the TRANSCEND trial [54] (Supplemental Table 2). Hypogammaglobulinemia inputs were based on the Institute for Clinical and Economic Review’s 2018 assessment of CAR T-cell therapies for B-cell cancers [52] (Supplemental Table 3). For all other grade 3–4 AEs, per-event costs were sourced from the HCUP [50] (Supplemental Table 4); management of grade 1–2 AEs only were assumed to require a single general practitioner visit.
Overall costs of monitoring were calculated from the unit costs for each resource and their frequencies (Supplemental Table 5). The types of resources and frequencies associated with the 28 days after infusion and PFS are modeled separately for CAR T-cell therapy and salvage chemotherapy (for those who failed to receive CAR T-cell infusion), to reflect different treatment-specific monitoring requirements. Monitoring frequencies are treatment-independent once patients remain in PFS for more than 2 years or enter a postprogression period.
Model Verification and Validation
Model programming underwent technical validation by a modeler not involved in its development. Additionally, model projections of PFS and OS for liso-cel were compared with the Kaplan-Meier data for the leukapheresed population in TRANSCEND to ensure model assumptions and survival analyses were valid.
Analyses
The model estimated incremental cost-effectiveness ratios using life-years (LY) and QALYs over a lifetime horizon; the incremental net monetary benefit (INMB) was calculated basis on a willingness-to-pay (WTP) threshold of $100,000 as the recommended lower bound by the Institute for Clinical and Economic Review [55]. Deterministic sensitivity analyses, probabilistic sensitivity analyses (PSA), and scenario analyses were conducted to explore the impact of uncertainty in model parameters and structural assumptions (Supplementary Material).
Scenario analyses were performed to test alternative settings and data sources for the model inputs (Supplemental Tables 6 and 7). Two key scenarios focused on differences between the CAR T-cell therapy trials. The first assumed that all patients received their CAR T-cell infusion as this may differ between clinical practice and the trials. The second explored the impact of potential bias resulting from a difference in the ZUMA-1 and TRANSCEND designs regarding bridging therapy use. This scenario compared liso-cel with axi-cel using an MAIC that matched on bridging (i.e., excluding patients from TRANSCEND who received bridging therapy).
Compliance with Ethics Guidelines
Data used in this analysis were derived from three previously conducted CAR T-cell therapy trials and does not contain any new studies on human participants or animals performed by any of the authors.
Results
Base-Case Analysis
Liso-cel Versus Axi-cel
The proportions of patients remaining alive at 5 years were 43% for liso-cel versus 39% for axi-cel (Fig. 2A). Liso-cel accrued marginally fewer LYs (liso-cel: 6.18 LYs; axi-cel: 6.22 LYs) but equivalent QALYs (liso-cel: 5.09; axi-cel: 5.09) because of a lower AE-related QALY decrement for liso-cel (Table 3). Total costs were higher for axi-cel ($515,085) than liso-cel ($440,106), primarily because of higher drug acquisition costs due to more patients receiving CAR T-cell infusion and AE management costs, which were more than three times as high for axi-cel than liso-cel ($51,643 vs $16,180). Liso-cel was less costly (incremental cost of − $74,980) and marginally more effective (0.002 incremental QALYs) than axi-cel, with a positive INMB of $75,170 at a WTP threshold of $100,000 per QALY gained (Table 3).
Liso-cel Versus Tisa-cel
A higher proportion of patients who received liso-cel remained alive at 5 years versus those who received tisa-cel (43% vs 25%) (Fig. 2b). Consequently, patients receiving liso-cel accrued greater LYs (6.18) and QALYs (5.09) compared with tisa-cel (3.75 LYs; 3.07 QALYs). Tisa-cel was associated with lower total costs than liso-cel ($372,180 vs $440,106), which was primarily due to the lower proportion of tisa-cel–treated patients receiving planned CAR T-cell infusion. Treatment with liso-cel was associated with an incremental cost of $67,925 and 2.02 incremental QALYs over tisa-cel, resulting in an incremental cost per QALY gained of $33,618 (Table 3).
Deterministic Sensitivity Analyses
Liso-cel Versus Axi-cel
The 15 parameters that resulted in the greatest change in the base-case INMB are presented in Fig. 3A. The key driver of model results was the OS cure fraction for liso-cel. Of the remaining parameters, the proportion of patients experiencing grade 3–4 AEs for axi-cel had the next largest impact on model results, but this impact was relatively small.

DSA tornado diagram showing incremental net monetary benefits: liso-cel versus axi-cel (A) and liso-cel versus tisa-cel (B). AE adverse event, axi-cel axicabtagene ciloleucel, CAR chimeric antigen receptor, CI confidence interval, CRS cytokine release syndrome, DSA deterministic sensitivity analysis, liso-cel lisocabtagene maraleucel, OS overall survival, PFS progression-free survival, tisa-cel tisagenlecleucel, USD United States dollars
Liso-cel Versus Tisa-cel
As in the comparison with axi-cel, the main driver of results versus tisa-cel was the OS cure fraction for liso-cel. In addition, the utility value for those in long-term remission and the proportion who failed to receive tisa-cel infusion had a moderate impact on INMB (Fig. 3B).
PSA
Liso-cel Versus Axi-cel
Results of the PSA simulations straddle the southwest and southeast quadrants, indicating certainty regarding cost savings with liso-cel (cheaper in almost all simulations) but uncertainty in incremental benefit (Fig. 4A). Half the simulations support the deterministic result that liso-cel was more effective and less costly than axi-cel. At a WTP threshold of $100,000 per QALY, liso-cel had an 82% probability of being cost-effective versus axi-cel (Fig. 4B).

Incremental cost-effectiveness scatter plot (A) and cost-effectiveness acceptability curve for liso-cel versus axi-cel (B) and liso-cel versus tisa-cel (C). axi-cel axicabtagene ciloleucel, liso-cel lisocabtagene maraleucel, tisa-cel tisagenlecleucel, QALY quality-adjusted life-year
Liso-cel Versus Tisa-cel
Most of the simulations for liso-cel versus tisa-cel (98%) fall within the northeast quadrant of the cost-effectiveness plane, supporting the deterministic results that liso-cel provides better health outcomes at a higher cost (Fig. 4A). At a WTP threshold of $100,000 per QALY, liso-cel had a 96% probability of being cost-effective versus tisa-cel (Fig. 4C).
Scenario Analysis
Liso-cel Versus Axi-cel
The results for the top 15 scenarios that resulted in the greatest change in the base-case INMB are presented in Supplemental Table 6. Key scenarios included naïve comparisons (instead of MAIC) for OS and PFS, assuming salvage chemotherapy efficacy for liso-cel–treated patients receiving nonconforming product, assuming all leukapheresed patients receive CAR T-cell therapy, and using an alternative source and costing approach for AEs.
Results are also presented for two scenarios that explored the impact of differences in the TRANSCEND and ZUMA-1 study designs. In the scenario that assumed all patients received CAR T-cell infusion, liso-cel was more effective and less costly than axi-cel, with greater QALYs and less cost-savings compared with the base case (Table 4). In the scenario using an MAIC versus axi-cel for PFS and OS that matched on bridging therapy, results were similar to the base case, with slightly increased LYs and QALYs and marginally greater cost savings for liso-cel versus axi-cel (Table 4).
Liso-cel Versus Tisa-cel
The scenarios resulting in the greatest change in the base-case INMB versus tisa-cel were variations in the discount rate for health outcomes and the model time horizon, naïve comparisons (instead of MAIC) for OS and PFS, and assuming all leukapheresed patients receive CAR T-cell therapy (Supplemental Table 7). In the latter, liso-cel remained highly cost-effective versus tisa-cel (Table 4).
Discussion
This analysis estimated that liso-cel is cost-effective compared with tisa-cel, yielding greater LYs and QALYs, with modest increases in costs. Compared with axi-cel, liso-cel yielded marginally fewer LYs but marginally greater QALYs at lower cost, owing to liso-cel’s favorable safety profile (resulting in lower QALY decrements and management costs due to AEs). Specifically, liso-cel is associated with lower rates of grade 3 or higher CRS and NEs than axi-cel based on TRANSCEND and ZUMA-1, respectively (2% CRS, 10% NEs for liso-cel [30] versus 11% CRS, 32% NEs for axi-cel [38], unadjusted rates). Compared with tisa-cel, liso-cel accrued incremental LY (2.43) and QALY (2.02) gains, with a large increase in incremental cost (+ $67,925); the lower cost of tisa-cel was driven primarily by the lower proportion of patients receiving CAR T-cell therapy than were planned. Results remained relatively consistent in sensitivity analyses, with the liso-cel OS cure fraction being the main driver of cost-effectiveness compared with both axi-cel and tisa-cel. Health outcomes (LYs and QALYs) of this analysis were similar to results reported in other published cost-effectiveness studies in the US as outlined in Supplemental Table 8 [19, 23, 27, 28, 52]. The ranges of axi-cel discounted LYs and QALYs, respectively, were 7.35–9.49 and 4.28–7.67; the only reported discounted LYs for tisa-cel were 6.73, and the range of QALYs was 2.82–5.16.
The model used a partitioned survival approach to estimate health state occupancy, which is commonly used in other published models of CAR T-cell therapies [13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28]. The design captured the unique stages of CAR T-cell therapy, including the pretreatment period. Extrapolation of survival outcomes using MCM has been widely accepted in health technology assessments of CAR T-cell therapies in R/R LBCL [15, 21, 22, 25, 56]. MCM also provided the best fit of the survival models assessed, and the resulting model PFS and OS projections for liso-cel (based on the infused cohort and pretreatment stratification to liso-cel and salvage chemotherapy) provided a very close fit to the Kaplan-Meier curves for the leukapheresed set in TRANSCEND.
CAR T-cell therapy represents a new treatment paradigm, and real-world data on patterns of use, safety, and effectiveness are still immature, which this analysis attempted to mitigate. Typical approaches for costing AE management (e.g., based on hospital stays) may underestimate resources required to manage CAR T-cell therapy–specific toxicities, such as CRS, NE, and hypogammaglobulinemia. Accordingly, the base-case analysis used a microcosting approach to estimate these costs. Additionally, the analysis used the longest follow-up from each trial available when the model was developed (TRANSCEND: 24 months [32]; ZUMA-1: 27.1–51.1 months [37, 38]; and JULIET: 40.3 months [39]), helping to minimize uncertainty in the MCM and estimated cure fractions. The analysis also leveraged the larger TRANSCEND patient population (n = 270) relative to the other trials and EQ-5D-5L data from TRANSCEND, wherever possible.
Without head-to-head studies, MAICs of effectiveness and safety for liso-cel versus axi-cel and tisa-cel were conducted to adjust for key differences between trial populations (eligibility criteria and baseline characteristics) and hence minimize potential bias induced when comparing single-arm trials [40, 41]. Although MAICs were conducted for efficacy and safety data, there were limitations to this analysis where not all differences in trial designs could be thoroughly adjusted for, which may bias cost-effectiveness. There were notable differences in the pivotal trial designs for each CAR T-cell therapy. These include patients’ disease histology during enrollment, CAR T-cell infusion setting (inpatient and/or outpatient), whether bridging therapy was permitted, time to CAR T-cell infusion, and whether patients had a confirmed manufacturing slot before enrollment [57].
Firstly, ZUMA-1 did not permit bridging therapy per protocol, and the MAIC used in the base case did not adjust for this difference [41], which may bias against liso-cel. Patients who received bridging therapy in TRANSCEND had worse prognoses, and the base-case results are likely to underestimate the value of liso-cel versus axi-cel. In the scenario analysis that adjusted for bridging therapy, liso-cel was more effective and less costly than axi-cel with similar incremental costs and QALYs to the base case.
Secondly, ZUMA-1 did not permit enrollment and leukapheresis unless a manufacturing slot was available, whereas TRANSCEND and JULIET enrolled patients before confirming a manufacturing slot [57]. This may have reduced the duration of the pretreatment period in ZUMA-1 and increased the percentage of patients who received their CAR T-cell infusion. In contrast, the pretreatment period was longer and more manufacturing failure was observed in JULIET than in ZUMA-1 and TRANSCEND. Furthermore, differences in the proportion of leukapheresed patients who received CAR T-cell infusions are a driver of cost-effectiveness, illustrated by the scenario analysis in which the incremental cost-effectiveness ratio versus tisa-cel was approximately half the base case ($14,352 vs $33,618) and liso-cel remained more effective and cost-saving versus axi-cel with a larger QALY gain (0.402 vs 0.002).
Although the analysis leveraged the TRANSCEND EQ-5D data wherever possible, there were limitations to the data. MMRM analysis indicated that utility for patients in PFS (without AEs) was marginally higher than that of the general population based on US index scores and age-adjusted population norms. This finding may demonstrate potential bias as TRANSCEND is a single-arm, open-label trial. Because TRANSCEND data could not inform all utility inputs owing to small numbers of events or lack of a signal from the data, sources using different elicitation methods or populations were used. This included a vignette study to inform the disutility of grade 3 or higher CRS, valued via time trade-off, which improves on data used in other cost-effectiveness analyses of CAR T-cell therapies [16, 17, 58, 59].
Clinical trials may not accurately reflect real-world practice. Additional data challenges necessitated modeling assumptions; for example, based on the assessment of patient-level data from TRANSCEND, efficacy was assumed to be the same for patients receiving liso-cel and nonconforming product, and because of a lack of reported data, 0% of patients in the axi-cel and tisa-cel arms were assumed to receive product outside of specifications. Protocol procedures in CAR T-cell trials have evolved regarding the use of prophylactic corticosteroids and earlier corticosteroid/tocilizumab treatment-emergent AE management, which may change the cost inputs in models such as ours [9, 60]. Additionally, bridging therapies before and subsequent therapies after CAR T-cell therapies have changed since the conduct of the pivotal studies included in this analysis, which may influence treatment outcomes and toxicities; current data are limited and are not expected to materially affect cost-effectiveness, as changes should affect all three CAR T-cell therapies equally. Finally, despite use of the longest follow-up available from TRANSCEND, ZUMA-1, and JULIET at the time of model development, longer follow-up from these trials and real-world cohorts would enable further validation of the long-term survival projections in this cost-effectiveness analysis.
Conclusions
This analysis estimated that liso-cel is cost-effective compared with axi-cel and tisa-cel from a US commercial payer perspective. Compared with axi-cel, liso-cel generated similar QALYs at lower cost, partly owing to lower rates of highly burdensome AESIs for CAR T-cell therapies. Compared with tisa-cel, liso-cel generated greater QALYs owing to superior survival, but at slightly higher cost because of higher drug acquisition cost. Overall, our findings provide the comparative cost-effectiveness of CAR T-cell therapies and highlight some of the key drivers of the cost-effectiveness (e.g., acquisition costs, AE rates, and survival benefits), which can inform clinical and payer decision-makers.
References
Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130:1800–8.
Raut LS, Chakrabarti PP. Management of relapsed-refractory diffuse large B cell lymphoma. South Asian J Cancer. 2014;3:66–70.
Tilly H, Gomes da Silva M, Vitolo U, et al. Diffuse large B-cell lymphoma (DLBCL): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26:v116–25.
Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125:22–32.
Zelenetz AD, Gordon LI, Abramson JS, et al. NCCN guidelines insights: B-cell lymphomas, version 3. J Natl Compr Canc Netw. 2019;17:650–61.
Kite Pharma. Yescarta (axicabtagene ciloleucel) suspension for intravenous infusion: U.S. prescribing information. 2022. https://www.fda.gov/media/108377/download. Accessed 20 Jan 2023.
Novartis. Kymriah (tisagenlecleucel) suspension for intravenous infusion: U.S. prescribing information. 2020. https://www.fda.gov/media/107296/download. Accessed 20 January 2023.
Bristol Myers Squibb. U.S. Food and Drug Administration approves Bristol Myers Squibb’s Breyanzi (lisocabtagene maraleucel), a new CAR T cell therapy for adults with relapsed or refractory large B-cell lymphoma. 2021. https://news.bms.com/news/details/2021/U.S.-Food-and-Drug-Administration-Approves-Bristol-Myers-Squibbs-Breyanzi-lisocabtagene-maraleucel-a-New-CAR-T-Cell-Therapy-for-Adults-with-Relapsed-or-Refractory-Large-B-cell-Lymphoma/default.aspx. Accessed 18 Apr 2022.
Iacoboni G, Villacampa G, Martinez-Cibrian N, et al. Real-world evidence of tisagenlecleucel for the treatment of relapsed or refractory large B-cell lymphoma. Cancer Med. 2021;10:3214–23.
Neelapu SS, Locke FL, Bartlett NL, et al. Comparison of 2-year outcomes with CAR T cells (ZUMA-1) vs salvage chemotherapy in refractory large B-cell lymphoma. Blood Adv. 2021;5:4149–55.
Salles G, Spin P, Liu FF, Garcia J, Kim Y, Hasskarl J. Indirect treatment comparison of liso-cel vs. salvage chemotherapy in diffuse large B-cell lymphoma: TRANSCEND vs. SCHOLAR-1. Adv Ther. 2021;38:3266–80.
Borgert R. Improving outcomes and mitigating costs associated with CAR T-cell therapy. Am J Manag Care. 2021;27:S253–61.
Balmaceda C, Abbott T, Espinoza MA. PCN92 Cost-effectiveness analysis of polatuzumab in combination with rituximab and bevacizumab for the treatment of patients with relapsed/refractory diffuse large B-cell lymphoma. Value Health. 2020;23(Suppl 1):S40.
Betts KA, Thuresson PO, Du EX, et al. PCN238 Is polatuzumab vedotin plus bendamustine-rituximab cost-effective for patients in the United States with transplant-ineligible relapsed/refractory diffuse large B-cell lymphoma? Value Health. 2019;22(Suppl 3):S482.
Canadian Agency for Drugs and Technologies in Health. CADTH optimal use report: axicabtagene ciloleucel for diffuse large B-cell lymphoma: economic review report. 2019. https://www.cadth.ca/sites/default/files/pdf/car-t/ct0002-axicabtagene-ciloleucel-economic-report-redacted.pdf. Accessed 18 Apr 2022.
Canadian Agency for Drugs and Technologies in Health. CADTH optimal use report: tisagenlecleucel for diffuse large B-cell lymphoma: economic review report. 2019. https://cadth.ca/sites/default/files/pdf/car-t/op0538-tisagenlecleucel-economic-report-DLBCL-jan2019.pdf. Accessed 18 Apr 2022.
Cher BP, Gan KY, Aziz MIA, et al. Cost utility analysis of tisagenlecleucel vs salvage chemotherapy in the treatment of relapsed/refractory diffuse large B-cell lymphoma from Singapore’s healthcare system perspective. J Med Econ. 2020;23:1321–9.
Karampampa K, Stene E, Axelsen F, et al. PPM3 Cost-effectiveness of axicabtagene ciloleucel (axi-cel) vs standard of care for adult patients with relapsed or refractory diffuse large B-cell lymphoma in Sweden, Norway, Finland, and Denmark. Value Health. 2020;23(Suppl 2):S687.
Liu R, Oluwole OO, Diakite I, Botteman MF, Snider JT, Locke FL. Cost effectiveness of axicabtagene ciloleucel versus tisagenlecleucel for adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy in the United States. J Med Econ. 2021;24:458–68.
Liu R, Snider JT, Diakite I, Tempelaar S, Botteman M. Cost effectiveness of axicabtagene ciloleucel (axi-cel) and tisagenlecleucel (tisa-cel) for adult patients with relapsed or refractory large B-cell lymphoma (RR LBCL) in the US. HemaSphere. 2020;4:794.
National Institute for Health and Care Excellence (NICE). Axicabtagene ciloleucel for treating diffuse large B-cell lymphoma and primary mediastinal large B-cell lymphoma after 2 or more systemic therapies: technology appraisal guidance [TA559]. 2019. https://www.nice.org.uk/guidance/ta559. Accessed 18 Apr 2022.
National Institute for Health and Care Excellence (NICE). Tisagenlecleucel for treating relapsed or refractory diffuse large B-cell lymphoma after 2 or more systemic therapies: technology appraisal guidance [TA567]. 2019. https://www.nice.org.uk/guidance/ta567. Accessed 18 Apr 2022.
Roth JA, Sullivan SD, Lin VW, et al. Cost-effectiveness of axicabtagene ciloleucel for adult patients with relapsed or refractory large B-cell lymphoma in the United States. J Med Econ. 2018;21:1238–45.
Scottish Medicines Consortium. Axicabtagene ciloleucel 0.4 – 2 × 108 cells dispersion for infusion dispersion for infusion (Yescarta®). SMC Advice on New Medicines 2019. https://www.scottishmedicines.org.uk/media/4121/axicabtagene-ciloleucel-yescarta-final-nov2018-for-website.pdf. Accessed 18 Apr 2022.
Scottish Medicines Consortium. Tisagenlecleucel 1.2 × 106 to 6 × 108 cells dispersion for infusion (Kymriah®). SMC Advice on New Medicines 2019. https://www.scottishmedicines.org.uk/media/4132/tisagenlecleucel-kymriah-final-jan-2019-for-website.pdf. Accessed 18 Apr 2022.
Sierra J, Briones J, Calleja MA, et al. Axicabtagene ciloleucel for the management of patients with diffuse large B-cell lymphoma and primary mediastinal large B-cell lymphoma: an economic evaluation for Spain. HemaSphere. 2020;4:810.
Whittington MD, McQueen RB, Ollendorf DA, et al. Long-term survival and cost-effectiveness associated with axicabtagene ciloleucel vs chemotherapy for treatment of B-cell lymphoma. JAMA Netw Open. 2019;2:e190035.
Qi CZ, Bollu V, Yang H, Dalal A, Zhang S, Zhang J. Cost-effectiveness analysis of tisagenlecleucel for the treatment of patients with relapsed or refractory diffuse large B-cell lymphoma in the United States. Clin Ther. 2021;43:1300–19.e8.
Caro JJ, Briggs AH, Siebert U, Kuntz KM, ISPOR-SMDM Modeling Good Research Practices Task Force. Modeling good research practices–overview: a report of the ISPOR-SMDM Modeling Good Research Practices Task Force-1. Med Decis Making. 2012;32:667–77.
Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839–52.
Sanders GD, Neumann PJ, Basu A, et al. Recommendations for conduct, methodological practices, and reporting of cost-effectiveness analyses: second panel on cost-effectiveness in health and medicine. JAMA. 2016;316:1093–103.
Abramson JS, Palomba ML, Gordon LI, et al. Two-year follow-up of TRANSCEND NHL 001, a multicenter phase 1 study of lisocabtagene maraleucel (liso-cel) in relapsed or refractory (R/R) large B-cell lymphomas (LBCL). Blood. 2021;138(Suppl 1):2840.
Jacobson C, Locke FL, Ghobadi A, et al. Long-term (≥4 year and ≥5 year) overall survival (OS) by 12- and 24-month event-free survival (EFS): an updated analysis of ZUMA-1, the pivotal study of axicabtagene ciloleucel (axi-cel) in patients (pts) with refractory large B-cell lymphoma (LBCL). Blood. 2021;138(Suppl 1):1764.
Schuster SJ, Tam CS, Borchmann P, et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021;22:1403–15.
National Institute for Health and Care Excellence (NICE). Flexible methods for survival analysis. 2020. https://www.sheffield.ac.uk/nice-dsu/tsds/flexible-methods-survival-analysis. Accessed 18 Apr 2022.
Rutherford MJ, Lambert PC, Sweeting MJ, et al. NICE DSU Technical Support Document 21. Flexible methods for survival analysis. 2020. http://www.nicedsu.org.uk. Accessed 20 Jan 2023.
Jacobson C, Locke FL, Ghobadi A, et al. Long-term survival and gradual recovery of B cells in patients with refractory large B cell lymphoma treated with axicabtagene ciloleucel (axi-cel). Blood. 2020;136:40–2.
Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20:31–42.
Jaeger U, Bishop MR, Salles G, et al. Myc expression and tumor-infiltrating T cells are associated with response in patients with relapsed/refractory diffuse large B-cell lymphoma (r/r DLBCL) treated with tisagenlecleucel in the JULIET trial. Blood. 2020;136:48–9.
Cartron G, Fox CP, Liu FF, et al. Matching-adjusted indirect treatment comparison of chimeric antigen receptor T-cell therapies for third-line or later treatment of relapsed or refractory large B-cell lymphoma: lisocabtagene maraleucel versus tisagenlecleucel. Exp Hematol Oncol. 2022;11:17.
Maloney DG, Kuruvilla J, Liu FF, et al. Matching-adjusted indirect treatment comparison of liso-cel versus axi-cel in relapsed or refractory large B cell lymphoma. J Hematol Oncol. 2021;14:140.
National Center for Health Statistics. National vital statistics report: United States life tables, 2017. 2019. https://www.cdc.gov/nchs/data/nvsr/nvsr68/nvsr68_07-508.pdf. Accessed 18 Apr 2022.
Maurer MJ, Ghesquières H, Jais JP, et al. Event-free survival at 24 months is a robust end point for disease-related outcome in diffuse large B-cell lymphoma treated with immunochemotherapy. J Clin Oncol. 2014;32:1066–73.
Adkins S. CAR T-cell therapy: adverse events and management. J Adv Pract Oncol. 2019;10(Suppl 3):21–8.
van Hout B, Janssen MF, Feng YS, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health. 2012;15:708–15.
Howell TA, Matza LS, Jun MP, Garcia J, Powers A, Maloney DG. Health state utilities for adverse events associated with chimeric antigen receptor T-cell therapy in large B-cell lymphoma. Pharmacoecon Open. 2022;6:367–76.
Juno Therapeutics. Breyanzi (lisocabtagene maraleucel) suspension for intravenous infusion: U.S. prescribing information. 2021. https://www.fda.gov/media/145711/download. Accessed 20 Jan 2023.
IBM. IBM Micromedex RED BOOK. 2020. https://www.ibm.com/products/micromedex-red-book. Accessed 18 Apr 2022.
Crump M, Kuruvilla J, Couban S, et al. Randomized comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine, and cisplatin chemotherapy before autologous stem-cell transplantation for relapsed and refractory aggressive lymphomas: NCIC-CTG LY.12. J Clin Oncol. 2014;32:3490–6.
Agency for Healthcare Research and Quality. Healthcare Cost and Utilization Project: weighted national estimates from HCUP National (Nationwide) Inpatient Sample (NIS), 2017; inflated to 2020. 2017. https://hcupnet.ahrq.gov/#setup. Accessed 18 Apr 2022.
Lin JK, Muffly LS, Spinner MA, Barnes JI, Owens DK, Goldhaber-Fiebert JD. Cost effectiveness of chimeric antigen receptor T-cell therapy in multiply relapsed or refractory adult large B-cell lymphoma. J Clin Oncol. 2019;37:2105–19.
Institute for Clinical and Economic Review. Chimeric antigen receptor T-cell therapy for B-cell cancers: effectiveness and value. 2018. https://collections.nlm.nih.gov/catalog/nlm:nlmuid-101744954-pdf. Accessed 18 Apr 2022.
Hettle R, Corbett M, Hinde S, et al. The assessment and appraisal of regenerative medicines and cell therapy products: an exploration of methods for review, economic evaluation and appraisal. Health Technol Assess. 2017;21:1–204.
Bristol Myers Squibb. Data on file. Liso-cel CRS and neurotoxicity analysis, BLA data cut USPI AE. 2021.
Institute for Clinical and Economic Review. 2020–2023 value assessment framework. 2020. https://icer.org/wp-content/uploads/2020/10/ICER_2020_2023_VAF_102220.pdf. Accessed 18 Apr 2022.
Scottish Medicines Consortium. Polatuzumab vedotin 140mg powder for concentrate for solution for infusion (Polivy®). 2020. https://www.scottishmedicines.org.uk/media/5360/polatuzumab-vedotin-polivy-final-august-2020-amended-180820-for-website.pdf. Accessed 18 Apr 2022.
Westin JR, Kersten MJ, Salles G, et al. Efficacy and safety of CD19-directed CAR-T cell therapies in patients with relapsed/refractory aggressive B-cell lymphomas: observations from the JULIET, ZUMA-1, and TRANSCEND trials. Am J Hematol. 2021;96:1295–312.
National Institute for Health and Care Excellence (NICE). Tisagenlecleucel-T for treating relapsed or refractory diffuse large B-cell lymphoma [ID1166]: committee papers. 2018. https://www.nice.org.uk/guidance/ta567/evidence/appraisal-consultation-committee-papers-pdf-6718510621. Accessed 18 Apr 2022.
National Institute for Health and Care Excellence (NICE). Axicabtagene ciloleucel for treating diffuse large B-cell lymphoma and primary mediastinal B-cell lymphoma after 2 or more systemic therapies [ID1115]: committee papers. 2018. https://www.nice.org.uk/guidance/ta559/evidence/appraisal-consultation-committee-papers-pdf-6661404973. Accessed 18 Apr 2022.
Riedell PA, Hwang WT, Nastoupil LJ, et al. Patterns of use, outcomes, and resource utilization among recipients of commercial axicabtagene ciloleucel and tisagenlecleucel for relapsed/refractory aggressive B cell lymphomas. Transplant Cell Ther. 2022;28:669–76.
Bristol Myers Squibb. Data on File. TRANSCEND. 19 June 2020 data cut-off.
Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380:45–56.
Matza LS, Cong Z, Chung K, et al. Utilities associated with subcutaneous injections and intravenous infusions for treatment of patients with bone metastases. Patient Prefer Adherence. 2013;7:855–65.
InHealth Professional Services. Physicians' fee & coding guide. Atlanta: InHealth Record Systems; 2019.
Bekelman JE, Halpern SD, Blankart CR, et al. Comparison of site of death, health care utilization, and hospital expenditures for patients dying with cancer in 7 developed countries. JAMA. 2016;315:272–83.
Sullivan PW, Ghushchyan V. Preference-based EQ-5D index scores for chronic conditions in the United States. Med Decis Making. 2006;26:410–20.
Acknowledgements
Funding
This study was funded by Bristol Myers Squibb. Publication fees including the journal’s Rapid Service and Open Access Fees were funded by Bristol Myers Squibb.
Medical Writing, Editorial, and Other Assistance
Writing and editorial assistance were provided by Jeremy Henriques, PhD, CMPP, and Tony Sica, PharmD, of The Lockwood Group (Stamford, CT, USA), funded by Bristol Myers Squibb.
Author Contributions
Christopher Parker, Fei Fei Liu, Kristen A. Deger, Conrado Franco-Villalobos, and Irina Proskorovsky contributed to the conception or design of the study; Conrado Franco-Villalobos and Irina Proskorovsky analyzed the data; and Christopher Parker, Fei Fei Liu, Kristen A. Deger, Conrado Franco-Villalobos, Irina Proskorovsky, Scott J. Keating, and Sonja Sorensen interpreted the data, contributed to the writing and reviewing of the manuscript, and approved the final manuscript.
Disclosures
Christopher Parker and Scott J. Keating were employees of and held stock in Bristol Myers Squibb at the time of this research; Christopher Parker is currently employed at Eisai EMEA and Scott J. Keating is employed at Vertex Pharmaceuticals. Fei Fei Liu is an employee of and holds stock in Bristol Myers Squibb. Kristen A. Deger, Conrado Franco-Villalobos, Irina Proskorovsky, and Sonja Sorensen are employees of Evidera, which was contracted by Bristol Myers Squibb to perform the study analysis.
Compliance with Ethics Guidelines
Data used in this analysis were derived from three previously conducted CAR T-cell therapy trials and does not contain any new studies on human participants or animals performed by any of the authors.
Data Availability
The datasets generated and/or analyzed during the current study for the TRANSCEND trial are patient-level data and are not publicly available due to patient privacy issues but are available upon reasonable request from Bristol Myers Squibb. Bristol Myers Squibb policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Parker, C., Liu, F.F., Deger, K.A. et al. Cost-Effectiveness of Lisocabtagene Maraleucel Versus Axicabtagene Ciloleucel and Tisagenlecleucel in the Third-Line or Later Treatment Setting for Relapsed or Refractory Large B-cell Lymphoma in the United States. Adv Ther 40, 2355–2374 (2023). https://doi.org/10.1007/s12325-023-02444-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-023-02444-x