|
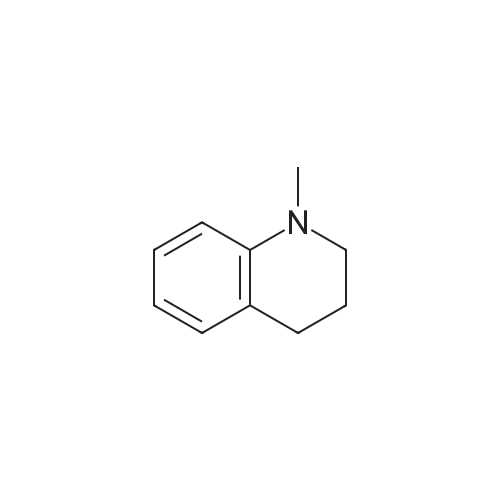
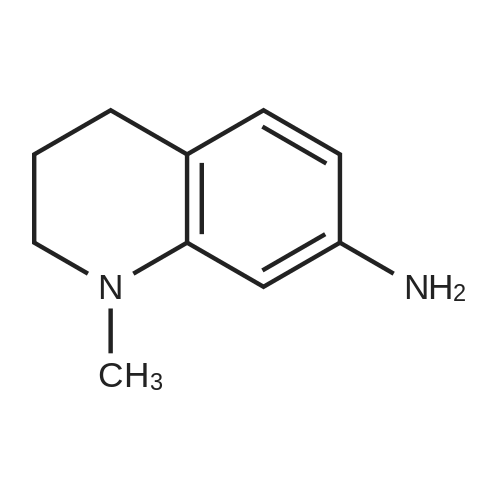
Stage #1: 1-methyl-7-nitro-1,2,3,4-tetrahydro-quinoline With hydrogenchloride; iron In water; N,N-dimethyl-formamide at 80 - 95℃; for 0.583333h;
Stage #2: With sodium hydrogencarbonate In water; N,N-dimethyl-formamide for 0.166667h; |
6.a
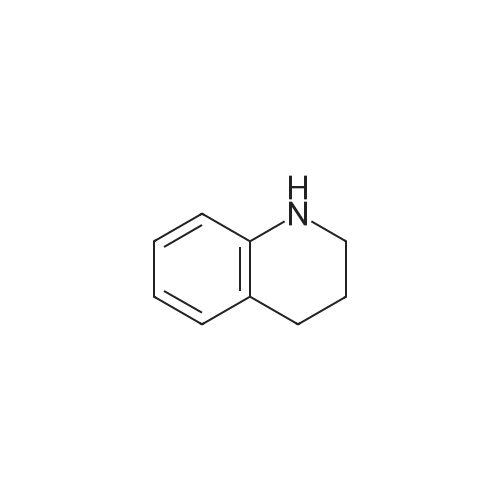
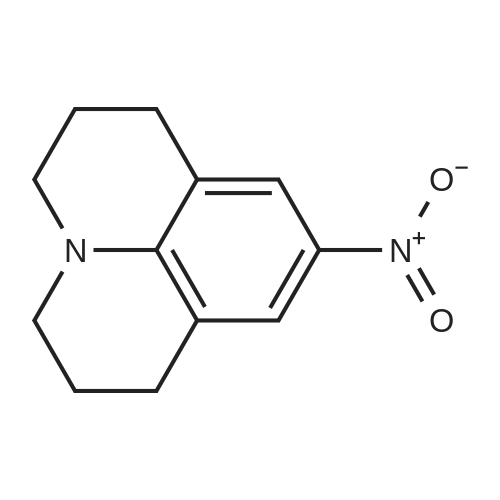
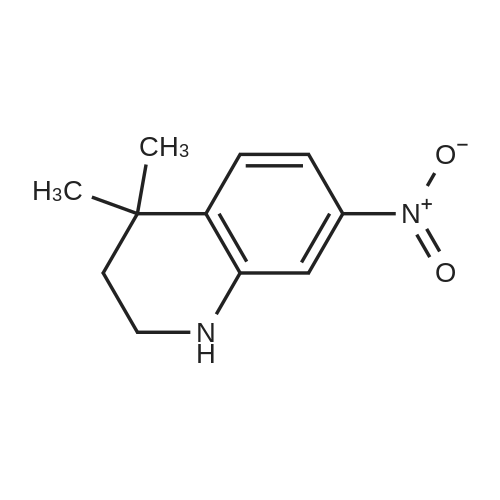
Here, 13.99 g of 1,2,3,4-tetrahydroquinoline and 14.52 g of potassium carbonate were dissolved into 350 ml of methanol. The resultant solution was maintained at temperatures between 45°C and 50°C with constant mixing. To this solution, 25.54 g of dimethyl sulfate was added dropwise. Subsequently, the solution was stirred for 3 hours at temperatures between 45°C and 50°C. Thereafter, the solution was left overnight. For toluene extraction, 350 ml of toluene and 350 ml of water were added to this solution. The extracted toluene solution was dried using anhydrous sodium sulfate, followed by removal of toluene to obtain a brown solution. This solution was column-purified to obtain 12.9. g of a pale yellow solution represented by the following structural formula (e). [Show Image] Here, 12.9 g of the compound represented by the above structural formula (e) was added dropwise to 230 g of concentrated sulfuric acid which had been cooled to between 0°C and 5°C, while maintaining the temperature at 0°C to 5°C. Subsequently, a mixture of 36 g of concentrated sulfuric acid and 9.0 g of concentrated nitric acid was added dropwise to the resultant solution while maintaining the temperature at 0°C to 5°C. After the mixture solution was added, the solution was returned to room temperature, followed by stirring for 2 hours. The reacted solution was poured into 300 ml of ice water for cooling. Then, the solution was adjusted to pH 9 by the addition of 50% aqueous sodium hydroxide while cooling the solution. After stirring for 1 hour, the deposited crystal was separated by filtration and dried. Thus, 12.4 g of red crystal represented by the following structural formula (f) was obtained. [Show Image] Here, 31.4 g of iron powder was suspended in 183 ml of DMF-water (2: 1) solution, and the solution was heated to between 85°C and 90°C with constant mixing. To this solution, a mixture obtained by mixing 6.7 ml of hydrochloric acid with 91.5 ml of DMF-water (2:1) solution was added dropwise. Subsequently, while maintaining the temperature at 85°C to 95°C, 183 ml of DMF solution containing 12.0 g of the compound represented by the above structural formula (f) was added dropwise to this solution by taking 15 minutes. The resultant solution was stirred for 20 minutes at the temperature between 80°C to 90°C. Thereafter, while the solution was left for cooling, 6.39 g of sodium hydrogencarbonate was added thereto, followed by stirring for 10 minutes. The solution was then filtrated to remove iron powder, and the filtrate was poured into 500 ml of ice water for toluene extraction. After the resultant solution was dried using anhydrous sodium sulfate, toluene was removed to obtain 5.47 g of a brown liquid represented by the following structural formula (g). [Show Image] Under nitrogen flow, 9.31 g of trifluoromethanesulfonic anhydride was stirred, and was maintained a temperature of 20°C or below. Here, 40 ml of toluene solution containing 5.47 g of the compound represented by the above structural formula (g) was added dropwise. The resultant solution was subsequently stirred for 4 hours at the temperature between 10°C and 15°C. Thereafter, the solution was left overnight. To the reacted solution, 2 ml of water is added at the temperature between 10°C to 25°C and was stirred for 1 hour. Thereafter, the deposited solid substance was the separated by filtration. The substance thus harvested was then dissolved into ethyl acetate, and 150 ml of water was added thereto. Then, the resultant solution was extracted with ethyl acetate. After the resultant solution was dried using anhydrous sodium sulfate, ethyl acetate was removed to obtain 6.87 g of a dark brown liquid represented by the following structural formula (h). [Show Image] [Show Image] Here, 0.58 g of 2-amino-5-methyl-1,3,4-thiadiazole represented by the above-described structural formula (i) was dissolved into a mixture of 5ml of acetic and 3 ml of propionic acid. To this solution, 1 ml of sulfuric acid was added dropwise at the temperature between 0°C and 5°C. Then, 1.78 g of 43% nitrosylsulfuric acid was added dropwise to the resultant solution at the temperature between 0°C and 5°C. In this way, diazotization was performed. Then, 1.77 g of the compound represented by the structural formula (h), 0.2 g of urea and 2.0 g of sodium acetate were dissolved into 20 ml of methanol. To this solution, the resultant diazo solution was added dropwise at the temperature between 0°C and 5°C and was stirred for 3 hours. Thereafter, the resultant solution was left overnight. The deposited crystal was separated by filtration and then dried. Thus, 1.44 g of red crystal was obtained which is represented by the following structural formula (j). [Show Image] Here, 1.30 g of an azo-compound represented by the structural formula (j), which was obtained as described above, was dissolved into 50 ml of THF. At room temperature, 6 ml of methanol solution containing 0.46 g of nickel acetate tetrahydrate was then added to the resultant solution. Thereafter, this solution was stirred for 3 hours at room temperature, followed by addition of 50 ml of water. The deposited crystal was then separated by filtration, and the obtained crystal was washed with water and dried. In this way, 0. 59 g of a nickel chelate compound was obtained. This compound showed λmax at 591 nm (ε=9.9 x 104) (in chloroform), 113L/gcm absorption coefficient per gram, and the coating film thereof showed the maximum absorption wavelength at 613 nm. (Measurement of the OD2/OD1 value) Here, 20 mg of dyes prepared in the examples 1 to 8 and comparative examples 1 to 6 were respectively added into 2 g of octafluoropentanol (OFP) solvents. Thereafter, supersonic dispersion was performed at temperatures between 50°C and 55°C for 60 minutes to obtain solutions A. The solutions A were then cooled to room temperature (25+/-5°C). Thereafter, the solutions A were filtrated through 0.2 µm filters (manufactured by Millipore Corporation) to obtain solutions B. The solutions B were respectively applied by spin coating on 1.2 mm thickness polycarbonate substrates having 170 nm groove depth, 500 nm groove width and 1600 nm trackpitch at a rotating speed of 800 rpm. Then, the thus obtained films composed of only dyes were annealed in the constant temperature oven for 5 minutes, where 80°C air is blowing. Thereafter, the substrates were left in a room, whereby the substrates were cooled to room temperature (fabrication of coated substrates A). The absorption spectra of these coated substrates A were then measured, wherein the coated substrates A were cut out to have sector shapes and were used as samples to be measured. Air was used as a reference sample. Measurement was then made using U-3300(manufactured by Hitachi, Ltd.). The following measurement conditions were adopted: wavelength scan speed of 300 nm/min; and optical density measuring (Absorbance) mode at a sampling cycle of 0.5 nm. Fig. 1 shows the absorption spectrum of the coated substrate A where the dye prepared in the example 1 is used. In addition, Table 1 shows wavelengths and optical densities at OD1 and OD2 as well as the OD2/OD1 value for each coated substrate A. Table 1 Number Film OD2 Film OD1 OD2/OD1 nm Abs nm Abs Example 1 607 0.815 559 0.64 1.273438 Example 2 609 0.805 560 0.62 1.298387 Example 3 607 0.796 558 0.618 1.288026 Example 4 610 0.744 559 0.583 1.276158 Example 5 608 0.797 559 0.617 1.291734 Example 6 608 0.782 559 0.604 1.294702 Example 7 609 0.815 560 0.637 1.279435 Example 8 613 0.754 556 0.589 1.280136 Comparative example 1 598 0.684 547 0.589 1.16129 Comparative example 2 604 0.66 555 0.533 1.238274 Comparative example 3 607 0.643 557 0.54 1.190741 Comparative example 4 609 0.696 557 0.597 1.165829' Comparative example 5 601 0.555 553 0.502 1.105578 Comparative example 6 613 0.625 562 0.533 1.172608 In addition, Fig. 3 shows the OD2/OD1 values measured in the examples 1 to 8 and in the comparative examples 1 to 6. As can be seen from the results shown in Fig. 3, there is a boundary between the values in the examples 1 to 8 and the values in the comparative examples 1 to 6 at 1.25. Moreover, Fig. 4 shows the maximum absorption wavelengths (wavelengths at which the OD2 values are obtained) of the coated substrates A fabricated in the examples 1 to 8 and in the comparative examples 1 to 5, and shows the measurement results for the reflectivity of each of the discs at a 40 mm radius, measured by using a DVD-ROM test system (647 nm). From the results shown in Fig. 4, it can be learned that the discs prepared in the comparative examples 1 to 5 tend to exhibit decreased reflectivity with increase in the maximum absorption wavelengths as indicated by the regression line in Fig. 4. As a trend in the dyes prepared in the comparative examples 1 to 5, it is difficult to obtain a reflectivity of 45% or more that is required in the DVD standards for reading by using dyes having the absorption maximum at wavelengths longer than 605 nm. On the other hand, it can be determined that the discs using the dyes prepared in the examples 1 to 8 allow to secure high reflectivity even when dyes having the absorption maximum at the vicinity of 610 nm are used. (Measurement of reference data) Here, 0.06 g of the azo-metal chelate dyes, respectively represented by the following structural formulae (S-1) to (S-6) were added to 5 g of OFP solvents, and were subject to supersonic dispersion at 50°C for 60 minutes. Thereafter, the solutions were left in a room, whereby the solutions were cooled to room temperature. The solutions were then filtrated through 0.2 µm filters (manufactured by Millipore Corporation). The resultant solutions were applied by spin coating on mirror-finished surface replicas (polycarbonate substrates with no guide grooves) so that substantially half area of each disc surface was covered. After drying, a reflecting layer was sputtered on a part of a recording layer formed by the spin-coating. The step height between the uncoated portion covered with the reflecting layer and the recording layer covered with the reflecting layer was measured by using a three-dimensional surface roughness meter (ZYGO: Maxim 5800, manufactured by Canon Inc.), whereby the film thickness was determined. In addition, using the automatic wavelength-scanning ellipsometer (MEL-30S, manufactured by JASCO Corporation), light with wavelengths between 580 and 650 nm was applied to the recording layer to which the reflecting layer was not applied, thereby measuring the reflectivity and the phase difference in variable-angle measurement mode. Then, with reference to the above-described film thickness, the combination of refraction index n and extinction coefficient k, which gives favorable convergence, was determined for each wavelength. Among the refraction indices for the wavelengths thus obtained, the refraction index that gives the maximum value is defined as n. Meanwhile, for each of the (S-1) to (S-6) dyes, a coated substrate A formed by use of the dye was fabricated and the absorption spectrum was measured as in the case of the above-described "method of measuring the OD2/OD1 value" Then the OD2/OD1 values were calculated. Table 2 and Fig. 2 show the thus determined OD2/OD1 values and n values (maximum refraction indices). Starting from the left side (i.e., from the smallest OD2/OD1 value to the largest one), data points respectively represent the values for the (S-1) to (S-6) dyes. [Show Image] [Show Image] Table 2 Dye OD2/OD1 n (max) S-1 0.83 2.32 S-2 0.85 2.5 S-3 1 2.8 S-4 1.03 2.9 S-5 1.16 3 S-6 1.21 3.2 |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping