

Genetic Diversity of Viral Populations Associated with Ananas Germplasm and Improvement of Virus Diagnostic Protocols
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and RNA Extraction
2.2. RNA-Sequencing and Viral Genome Assembly
2.3. HTS Validation
2.4. Identification of a Novel Species within the Sadwavirus Genus from Public Domain Ananas spp. Transcriptome Datasets
2.5. Study of Intra-Species Recombination and Genomic Diversity
2.6. Phylogenetic Analyses to Explore Inter- and Intra-Species Relationships
2.7. Evaluating and Optimizing RT-PCR Assays for Sensitive Detection of Viruses Infecting Pineapple
3. Results
3.1. Characterization of the Virome in Pineapple Germplasm Allows the Discovery of Two Novel Sadwavirus Species
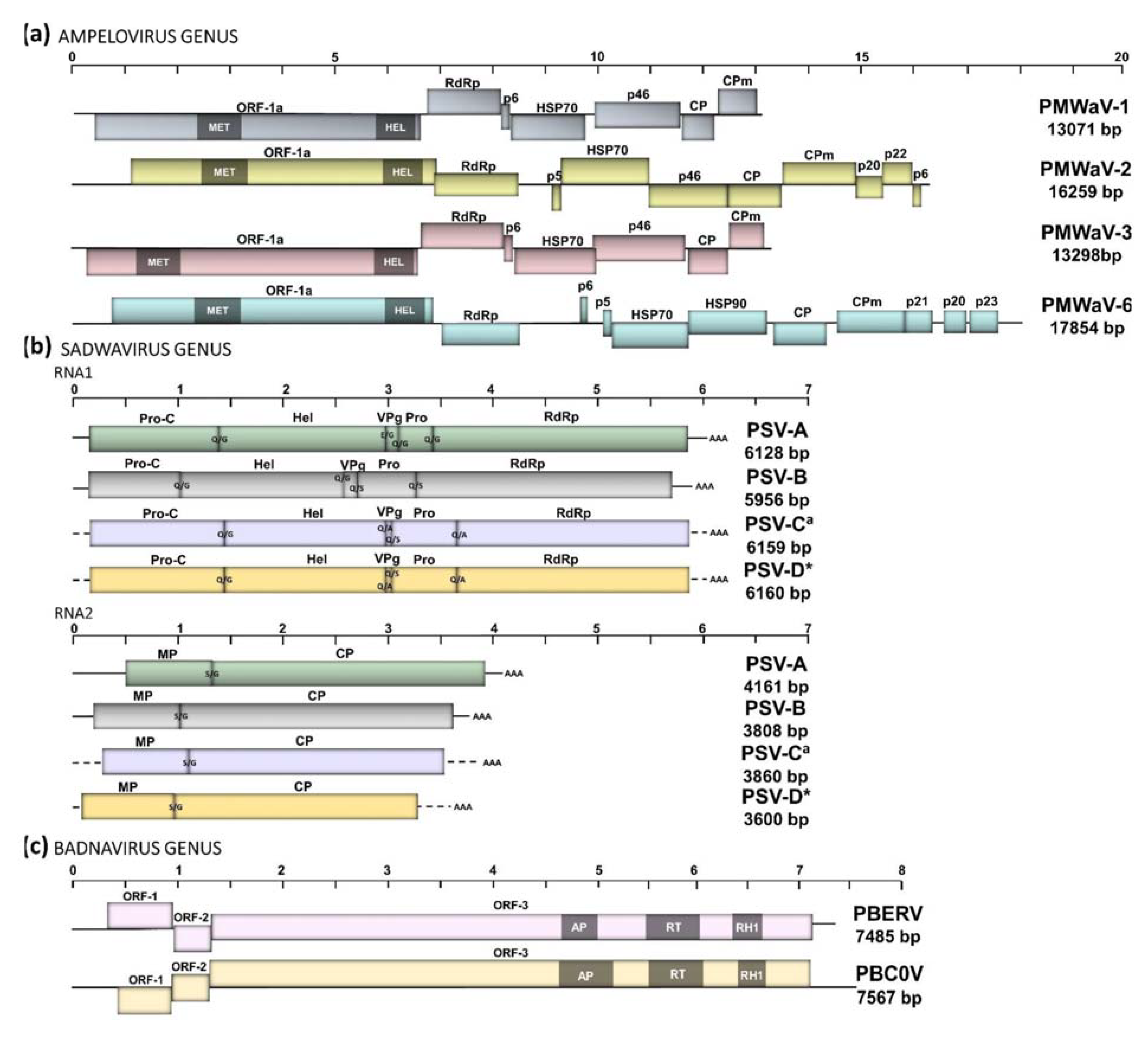
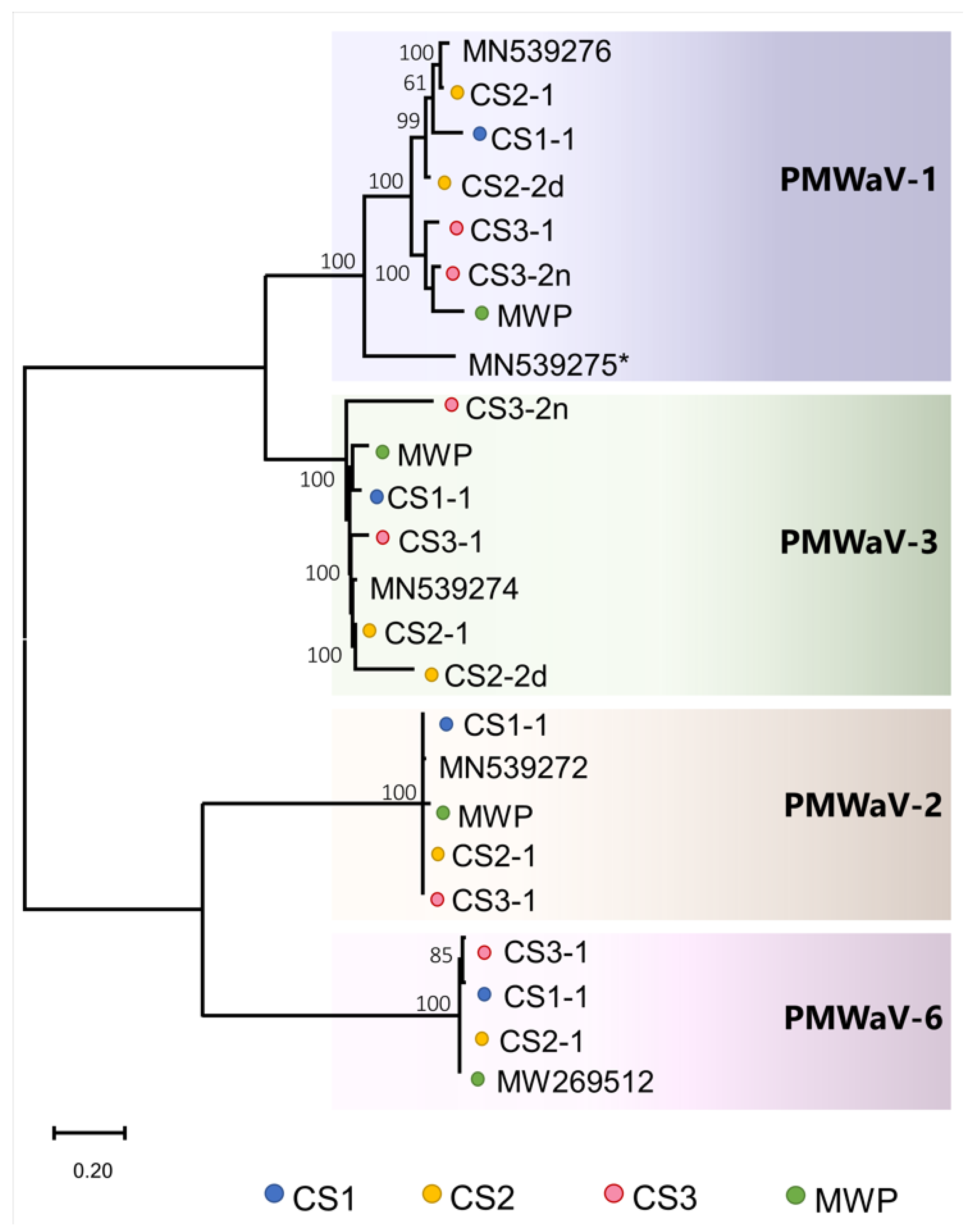
3.1.1. Ampeloviruses
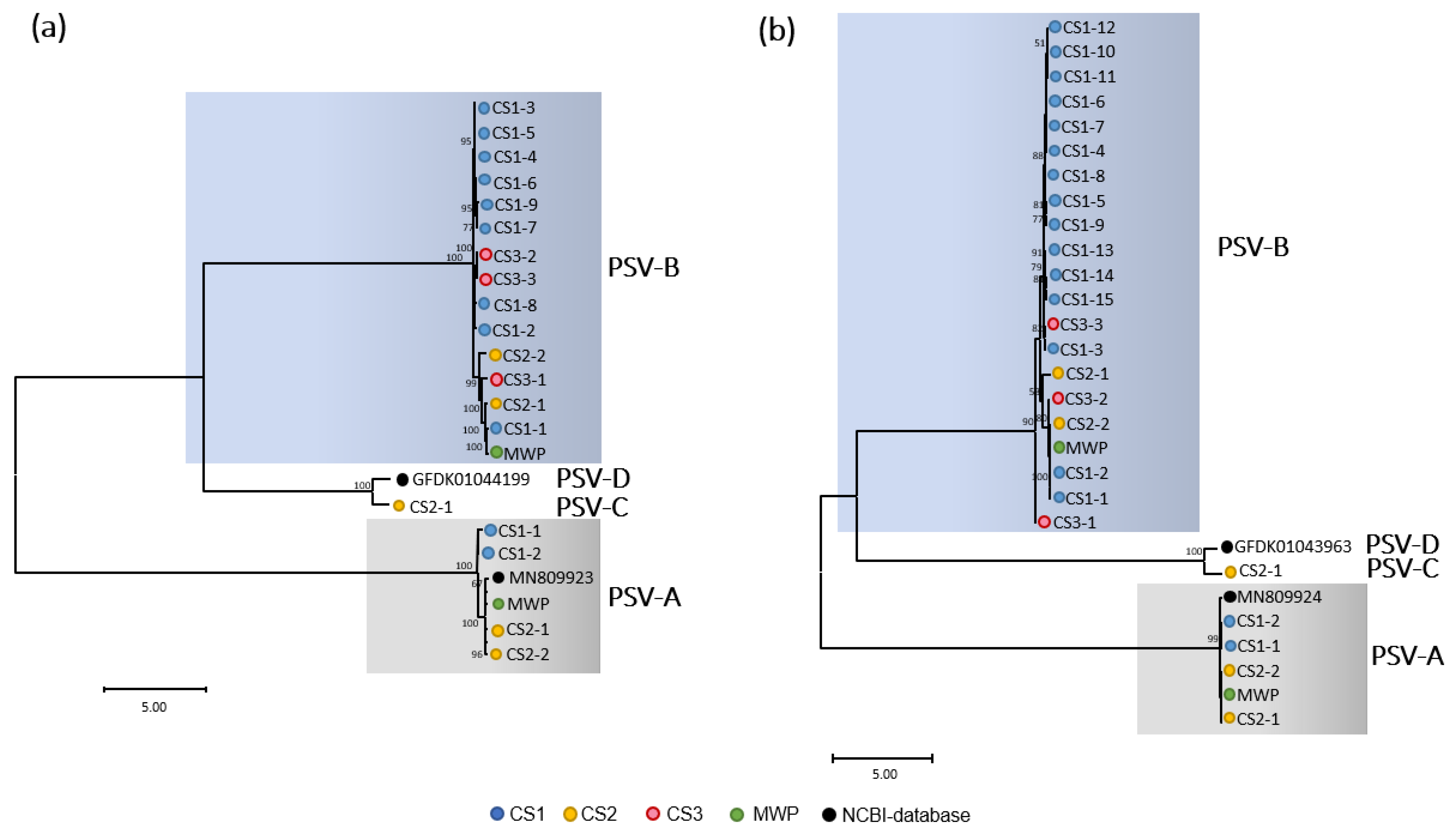
3.1.2. Sadwaviruses: Two Novel Species
3.1.3. Badnaviruses: Report of the Complete Genome of PBERV
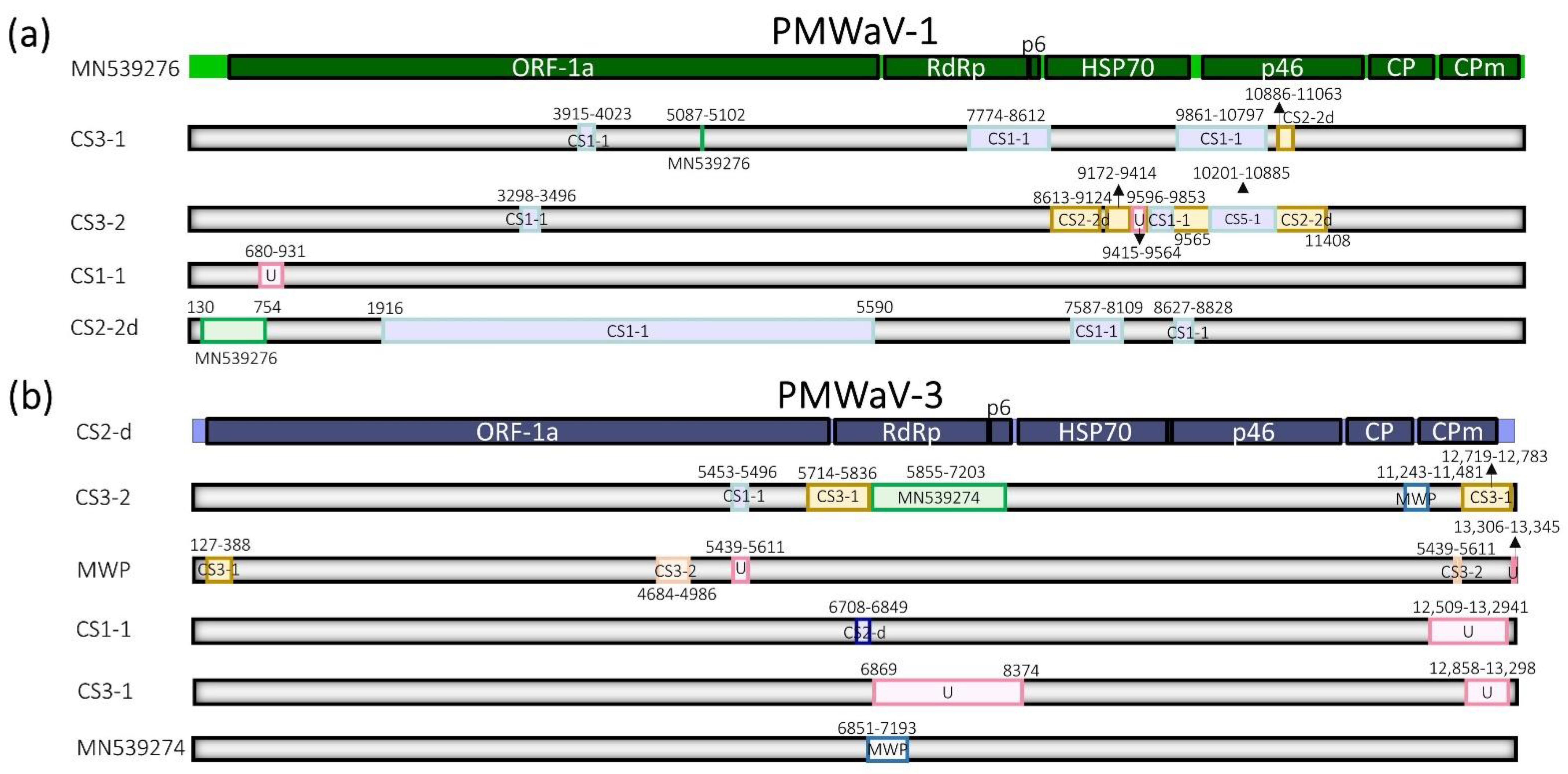
3.2. Inter- and Intra-Species Diversity and Recombination in Members of the PMWaV Complex
3.3. Recombination Drives Diversity and Evolution of Sadwaviruses Infecting Ananas spp.
3.4. Improvement of RT-PCR Detection Methods and Virus Indexing of Ananas Germplasm
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FAO. Food and Agriculture Organization of the United Nations. Available online: http://www.fao.org/land-water/databases-and-software/crop-information/pineapple/en/ (accessed on 23 September 2022).
- Dey, K.; Green, J.; Melzer, M.; Borth, W.; Hu, J. Mealybug Wilt of Pineapple and Associated Viruses. Horticulturae 2018, 4, 52. [Google Scholar] [CrossRef] [Green Version]
- Larrea-Sarmiento, A.; Olmedo-Velarde, A.; Green, J.C.; Al Rwahnih, M.; Wang, X.; Li, Y.H.; Wu, W.; Zhang, J.; Matsumoto, T.K.; Suzuki, J.Y.; et al. Identification and complete genomic sequence of a novel sadwavirus discovered in pineapple (Ananas comosus). Arch. Virol. 2020, 165, 1245–1248. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.J.; Karasev, A.V.; Sether, D.M.; Hu, J.S. Nucleotide sequence, genome organization and phylogenetic analysis of pineapple mealybug wilt-associated virus-2. J. Gen. Virol. 2001, 82, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.J.; Sether, D.M.; Karasev, A.V.; Borth, W.; Hu, J.S. Complete nucleotide sequence and genome organization of pineapple mealybug wilt-associated virus-1. Arch. Virol. 2008, 153, 707–714. [Google Scholar] [CrossRef]
- Sether, D.M.; Melzer, M.J.; Borth, W.B.; Hu, J.S. Genome organization and phylogenetic relationship of Pineapple mealybug wilt associated virus-3 with family Closteroviridae members. Virus Genes 2009, 38, 414–420. [Google Scholar] [CrossRef]
- Sether, D.M.; Hu, J.S. Closterovirus infection and mealybug exposure are necessary for the development of mealybug wilt of pineapple disease. Phytopathology 2002, 92, 928–935. [Google Scholar] [CrossRef] [Green Version]
- Sether, D.M.; Melzer, M.J.; Busto, J.; Zee, F.; Hu, J.S. Diversity and Mealybug Transmissibility of Ampeloviruses in Pineapple. Plant Dis. 2005, 89, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Larrea-Sarmiento, A.; Olmedo-Velarde, A.; Wang, X.; Borth, W.; Matsumoto, T.K.; Suzuki, J.Y.; Wall, M.M.; Melzer, M.; Hu, J. A novel ampelovirus associated with mealybug wilt of pineapple (Ananas comosus). Virus Genes 2021, 57, 464–468. [Google Scholar] [CrossRef]
- Subere, C.V.Q.; Sether, D.M.; Borth, W.B.; Melzer, M.J.; Hu, J.S. Transmission Characteristics of Pineapple Mealybug Wilt Associated Virus-2 by the Grey Pineapple Mealybugs Dysmicoccus Neobrevipes in Hawaii. 2011. Available online: https://www.actahort.org/books/902/902_47.htm (accessed on 25 October 2022).
- Gambley, C.F.; Geering, A.D.W.; Steele, V.; Thomas, J.E. Identification of viral and non-viral reverse transcribing elements in pineapple (Ananas comosus), including members of two new badnavirus species. Arch. Virol. 2008, 153, 1599–1604. [Google Scholar] [CrossRef]
- Hernandez-Rodriguez, L.; Ramos-Gonzalez, P.L.; Garcia-Garcia, G.; Javer Higginson, E.; Zamora-Rodriguez, V. First report of Pineapple bacilliform comosus virus (PBCoV) and endogenous Pineapple pararetrovirus-1 (ePPRV-1) in pineapple plants in Cuba. New Dis. Rep. 2013, 28, 2044-0588. [Google Scholar] [CrossRef]
- Larrea-Sarmiento, A.; Geering, A.D.W.; Olmedo-Velarde, A.; Wang, X.; Borth, W.; Matsumoto, T.K.; Suzuki, J.Y.; Wall, M.M.; Melzer, M.; Moyle, R.; et al. Genome sequence of pineapple secovirus B, a second sadwavirus reported infecting Ananas comosus. Arch. Virol. 2022, 2022, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Gambley, C.F.; Steele, V.; Geering, A.D.W.; Thomas, J.E. The genetic diversity of ampeloviruses in Australian pineapples and their association with mealybug wilt disease. Australas. Plant Pathol. 2008, 37, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Sether, D.M.; Melzer, M.J.; Borth, W.B.; Hu, J.S. Pineapple bacilliform CO virus: Diversity, Detection, Distribution, and Transmission. Plant Dis. 2012, 96, 1798–1804. [Google Scholar] [CrossRef] [Green Version]
- Green, J.C.; Rwahnih, M.A.; Olmedo-Velarde, A.; Melzer, M.J.; Hamim, I.; Borth, W.B.; Brower, T.M.; Wall, M.; Hu, J.S. Further genomic characterization of pineapple mealybug wilt-associated viruses using high-throughput sequencing. Trop. Plant Pathol. 2020, 45, 64–72. [Google Scholar] [CrossRef]
- Hernandez-Rodriguez, L.; Ramos-Gonzalez, P.; Garcia-Garcia, G.; Zamora, V.; Peralta-Martin, A.; Peña, I.; Perez, J.; Ferriol, X. Geographic distribution of mealybug wilt disease of pineapple and genetic diversity of viruses infecting pineapple in Cuba. Crop Prot. 2014, 65, 43–50. [Google Scholar] [CrossRef]
- Ho, T.; Martin, R.R.; Tzanetakis, I.E. Next-Generation Sequencing of Elite Berry Germplasm and Data Analysis Using a Bioinformatics Pipeline for Virus Detection and Discovery. In Plant Pathology: Techniques and Protocols; Lacomme, C., Ed.; Springer: New York, NY, USA, 2015; pp. 301–313. [Google Scholar]
- Diaz-Lara, A.; Stevens, K.A.; Klaassen, V.; Hwang, M.S.; Al Rwahnih, M. Sequencing a Strawberry Germplasm Collection Reveals New Viral Genetic Diversity and the Basis for New RT-qPCR Assays. Viruses 2021, 13, 1442. [Google Scholar] [CrossRef]
- Diaz-Lara, A.; Klaassen, V.; Stevens, K.; Sudarshana, M.R.; Rowhani, A.; Maree, H.J.; Chooi, K.M.; Blouin, A.G.; Habili, N.; Song, Y.; et al. Characterization of grapevine leafroll-associated virus 3 genetic variants and application towards RT-qPCR assay design. PLoS ONE 2018, 13, e0208862. [Google Scholar] [CrossRef] [Green Version]
- Nuzzo, F.; Moine, A.; Nerva, L.; Pagliarani, C.; Perrone, I.; Boccacci, P.; Gribaudo, I.; Chitarra, W.; Gambino, G. Grapevine virome and production of healthy plants by somatic embryogenesis. Microb. Biotechnol. 2022, 15, 1357–1373. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Rowhani, A.; Westrick, N.; Stevens, K.; Diaz-Lara, A.; Trouillas, F.P.; Preece, J.; Kallsen, C.; Farrar, K.; Golino, D. Discovery of Viruses and Virus-Like Pathogens in Pistachio using High-Throughput Sequencing. Plant Dis. 2018, 102, 1419–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 2019, 8, giz100. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longdon, B.; Murray, G.G.R.; Palmer, W.J.; Day, J.P.; Parker, D.J.; Welch, J.J.; Obbard, D.J.; Jiggins, F.M. The evolution, diversity, and host associations of rhabdoviruses. Virus Evol. 2015, 1, vev014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejerman, N.; Dietzgen, R.G.; Debat, H. Illuminating the Plant Rhabdovirus Landscape through Metatranscriptomics Data. Viruses 2021, 13, 1304. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Arif, M.; Ochoa-Corona, F.M. Comparative assessment of 5′ A/T-rich overhang sequences with optimal and sub-optimal primers to increase PCR yields and sensitivity. Mol. Biotechnol. 2013, 55, 17–26. [Google Scholar] [CrossRef]
- Gadberry, M.D.; Malcomber, S.T.; Doust, A.N.; Kellogg, E.A. Primaclade—A flexible tool to find conserved PCR primers across multiple species. Bioinformatics 2004, 21, 1263–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, K.K.; Borth, W.B.; Melzer, M.J.; Wang, M.L.; Hu, J.S. Analysis of pineapple mealybug wilt associated virus-1 and -2 for potential RNA silencing suppressors and pathogenicity factors. Viruses 2015, 7, 969–995. [Google Scholar] [CrossRef] [Green Version]
- Hohn, T.; Rothnie, H. Plant pararetroviruses: Rreplication and expression. Curr. Opin. Virol. 2013, 3, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Adiputra, J.; Jarugula, S.; Naidu, R.A. Intra-species recombination among strains of the ampelovirus Grapevine leafroll-associated virus 4. Virol. J. 2019, 16, 139. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, S.; Wu, J.; Yang, F.; Zhou, Y.; Zhou, C.; Cao, M. Complete genome sequences and recombination analysis of three divergent Satsuma dwarf virus isolates. Trop. Plant Pathol. 2021, 46, 26–30. [Google Scholar] [CrossRef]
- Satyanarayana, T.; Gowda, S.; Boyko, V.P.; Albiach-Marti, M.R.; Mawassi, M.; Navas-Castillo, J.; Karasev, A.V.; Dolja, V.; Hilf, M.E.; Lewandowski, D.J.; et al. An engineered closterovirus RNA replicon and analysis of heterologous terminal sequences for replication. Proc. Natl. Acad. Sci. USA 1999, 96, 7433–7438. [Google Scholar] [CrossRef] [Green Version]
- Mongkolsiriwattana, C.; Chen, A.Y.; Ng, J.C. Replication of Lettuce chlorosis virus (LCV), a crinivirus in the family Closteroviridae, is accompanied by the production of LCV RNA 1-derived novel RNAs. Virology 2011, 420, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Menzel, W.; Goetz, R.; Lesemann, D.E.; Vetten, H.J. Molecular characterization of a closterovirus from carrot and its identification as a German isolate of Carrot yellow leaf virus. Arch. Virol. 2009, 154, 1343–1347. [Google Scholar] [CrossRef]
- Mawassi, M.; Bar-Joseph, M. The defective RNAs of Closteroviridae. Front. Microbiol. 2013, 4, 132. [Google Scholar] [CrossRef]
- Burger, J.T.; Maree, H.J.; Gouveia, P.; Naidu, R.A. Grapevine leafroll-associated virus3. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Meng, B., Martelli, G.P., Golino, D.A., Fuchs, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 167–195. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Genus | Virus Specie | Target | Sequence 5′–3′ | Product Size (bp) | Length (bp) | Tm | Annealing PCR-Conditions a | Reference |
|---|---|---|---|---|---|---|---|---|
| Ampelovirus | PMWaV-1 | HSP70 | GGTTGTTCTATAGGACCAGT | 314 | 20 | 58 | 51 °C | This study |
| GTRCCYCCTCCGAAATCGTA | 20 | 60 | ||||||
| PMWaV-2 | CGAACTAGACTCATACGTGC | 571 | 20 | 50 | 55 °C | This study | ||
| CGGCTCATTAGTCACCTCCT | 20 | 55 | ||||||
| PMWaV-3 | ACDTTGTGTTTCTCCCCTGG | 272 | 20 | 60 | 51 °C | This study | ||
| GATCCATCRACGGGACCRAT | 20 | 60 | ||||||
| PMWaV-5 | CCCGGACGTGAATGATGAAG | 672 | 20 | 55 | 50 °C | This study | ||
| GTATTAGCTGCGCCCGTTCT | 20 | 55 | ||||||
| PMWaV-6 | GGAGTTGCGGGTTGTCTTCC | 450 | 20 | 64 | 55 °C | Larrea-Sarmiento et al., 2021 | ||
| ATAGCCCTCACCGGTACTCC | 20 | 64 | ||||||
| Sadwavirus | PSV-A RNA1 | RdRp | GTGACATTTTTCACRAAYTGGGA | 659 | 23 | 55 | 48 °C | This study |
| CTCTGCTGRCAYTGAGCAA | 19 | 56 | ||||||
| PSV-A RNA2 | CP | GAATCGCGYAATATGGTGCA | 544 | 20 | 58 | This study | ||
| ATCCTCCTGAGTCAGTRGGCAA | 22 | 59 | ||||||
| PSV-B RNA1 | RdRp | TAYAACTGGGAYATYATGGCDTC | 317 | 23 | 56 | 51 °C | This study | |
| CGCATTATDGCATACCARCT | 20 | 56 | ||||||
| PSV-B RNA2 | CP | AGVATGGGAGCKTTCCACAT | 415 | 20 | 58 | This study | ||
| GTGCANGARCCAGCTATRCT | 20 | 58 | ||||||
| PSVC-RNA1 | RdRp | CTGCTACCCAACCACCAGAA | 480 | 20 | 62 | 55 °C | This study | |
| CCATCACGGTCAAAGCAAATCC | 22 | 58 | ||||||
| PSVC-RNA2 | CP | GTCTGTTGTCTCTGCCCTCA | 539 | 20 | 62 | This study | ||
| TAYAAAGAACAACTGCCAGCAAC | 23 | 56 | ||||||
| PSVD-RNA1 | RdRp | GAGAGACGGGAACAGGAACC | 360 | 20 | 64 | 55 °C | This study | |
| TGCCCATCGGTAAGTCTCAC | 20 | 62 | ||||||
| PSVD-RNA2 | CP | GCTAAATCCCGTTGTGAAGCC | 657 | 21 | 58 | 51 °C | This study | |
| GCAACAGAAAGCTTGGAGTGG | 21 | 58 | ||||||
| Badnavirus | PBCOV | RNaseH | ACAAGGACCTTCAAACWACA | 631 | 20 | 56 | 51 °C | This study |
| TCTTCATGCAGCATCATBAC | 20 | 56 | ||||||
| PBERV | RNaseH | GGAGTGAAGCAGGTGGACTT | 463 | 20 | 62 | 55 °C | This study | |
| TYTCTGCGTCGATTGTGCTT | 20 | 58 |
| PMWaV | PSV | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample No. | HANA Accession | Origin | ID | -1 | -2 | -3 | -6 | -A | -B | -C | -D | PBCOV | PBERV | Summary |
| 1 | 6 | Taiwan 2 | Tainung #9 | + | + | + | − | + | + | − | − | − | − | 1, 2, 3, A, B |
| 2 | 12 | DR. Congo | Congo | + | + | + | − | + | + | − | − | + | − | 1, 2, 3, A, B, Co |
| 3 | 13 | American Samoa | Spanish Samoa | + | + | + | − | + | + | − | − | + | − | 1, 2, 3, A, B, Co |
| 4 | 14 | Singapur | Pernambuco | + | + | + | − | + | + | − | − | − | + | 1, 2, 3, A, B, Er |
| 5 | 16 | Barbados 2 | Bermuda | − | + | + | − | + | − | − | − | + | + | 2, 3, A, Co, Er |
| 6 | 17 | South Africa 3 | Natal | + | + | + | − | − | + | − | − | + | − | 1, 2, 3, A, B, Co |
| 7 | 18 | Taiwan | Mauritius | + | − | − | − | + | + | − | − | + | − | 1, A, B, Co |
| 8 | 20 | Brazil | F 101 | + | + | + | − | − | − | − | − | − | + | 1, 2, 3, Er |
| 9 | 21 | Brazil 1 | Abacaxi | + | + | + | − | − | − | − | − | − | − | 1, 2, 3 |
| 10 | 22 | Colombia | Santa Marta No. 1 | + | + | + | − | + | − | − | − | + | + | 1, 2, 3, A, Co, Er |
| 11 | 23 | Jamaica 2 | Sam Clarke | + | + | − | − | + | − | − | − | + | + | 1, 2, A, Co, Er |
| 12 | 27 | USA: HI, Oahu | Wild Kailua | + | + | + | − | − | + | − | − | − | − | 1, 2, 3, B |
| 13 | 31 | Philippines 2 | Black Antigua | + | + | − | − | − | − | − | − | − | + | 1, 2, Er |
| 14 | 33 | Indonesia 2 | Kendal | + | − | − | − | − | + | + | − | − | − | 1, B, C |
| 15 | 34 | Guatemala 1 | Monte Lirio | − | + | − | − | + | − | − | − | − | + | 2, A, Er |
| 16 | 35 | India 2 | Amalsad | + | − | − | − | − | − | + | − | − | − | 1, C |
| 17 | 36 | Panama | Cowboy | − | − | − | − | + | + | − | − | − | + | A, B, Er |
| 18 | 37 | Mexico 1 | Criolla | + | + | − | − | + | − | − | − | − | − | 1, 2, A |
| 19 | 38 | Brazil | Wild Brazil | − | − | + | − | − | − | − | − | − | − | 3 |
| 20 | 39 | Philippines | Philippine Hybrid | − | + | − | − | + | − | − | − | − | − | 2, A |
| 21 | 41 | Vietnam | Pho Lang Tuang | + | + | − | − | − | − | − | − | − | − | 1, 2 |
| 22 | 46 | Zaire 3 | Sugarloaf | + | + | + | + | − | − | − | − | − | − | 1, 2, 3, 6 |
| 23 | 48 | Mexico | Mexican Criolla | + | + | − | − | − | − | − | − | + | − | 1, 2, Co |
| 24 | 49 | Colombia 1 | Pina Criolla | + | + | + | − | − | + | − | − | − | + | 1, 2, 3, B, Er |
| 25 | 52 | Colombia | Papuri Vaupes Colombia | + | + | + | − | − | − | − | − | − | − | 1, 2, 3 |
| 26 | 53 | Samoa 3 | British Samoa P1 | + | − | + | − | − | − | − | − | + | − | 1, 3 |
| 27 | 54 | Western Samoa | British Samoa P5 | + | − | + | − | − | − | − | − | − | + | 1, 3, Er |
| 28 | 60 | Guatemala | Spanish Guatemala | + | + | + | − | − | + | − | − | − | − | 1, 2, 3, B |
| 29 | 63 | Brazil | CB 2 | − | + | − | − | + | − | − | − | + | + | 2, A, Co, Er |
| 30 | 64 | Brazil | CB 5 | − | − | − | − | − | − | − | − | − | − | - |
| 31 | 65 | Brazil | CB 6 | − | + | + | − | + | − | − | − | + | + | 2, 3, A, Co, Er |
| 32 | 67 | Brazil | CB 10 | + | + | − | − | − | − | − | − | − | − | 1, 2 |
| 33 | 68 | Brazil | CB 11 | − | − | − | − | − | − | − | − | − | + | Er |
| 34 | 69 | Paraguay | CB 15 | + | + | − | − | − | − | − | − | − | − | 1, 2 |
| 35 | 70 | Brazil | CB 17 | − | + | + | − | − | − | − | − | − | − | 2, 3 |
| 36 | 71 | Paraguay 1 | CB 18 | − | − | − | − | − | − | − | − | − | + | Er |
| 37 | 72 | Paraguay | CB 19 | − | − | − | − | + | − | − | − | − | − | A |
| 38 | 73 | Paraguay | CB 20 | − | − | − | − | − | + | − | − | + | − | B, Co |
| 39 | 74 | Paraguay | CB 21 | + | + | + | + | − | − | − | − | + | − | 1, 2, 3, 6, Co |
| 40 | 75 | Argentina | CB 23 | + | + | + | − | + | − | − | − | − | − | 1, 2, 3, A |
| 41 | 88 | Brazil | CB 71 | − | − | - | − | + | − | − | − | + | − | A, Co |
| 42 | 90 | Brazil 1 | Prazeres | − | + | + | + | + | + | − | − | + | + | 2, 3, 6, A, B, Co, Er |
| 43 | 91 | Trinidad 1 | Trinidad | − | - | − | − | − | − | − | − | − | − | - |
| 44 | 92 | USA: HI 1 | Cayenne 666 | + | + | − | − | + | − | − | − | + | + | 1, 2, A, Co, Er |
| 45 | 101 | USA: HI, Lanai | Cayenne M 4 W | + | + | − | − | + | − | − | − | + | − | 1, 2, A, Co |
| 46 | 110 | USA 2 | Cayenne M 109-5 | + | + | + | − | + | − | − | − | + | + | 1, 2, 3, A, Co, Er |
| 47 | 115 | USA: HI, Oahu | Cayenne M 226 Nubby | + | + | + | + | + | − | − | − | + | + | 1, 2, 3, 6, A, Co, Er |
| 48 | 117 | USA: HI, Maui 2 | Cayenne M 35 | + | + | − | − | + | − | − | − | + | + | 1, 2, A, Co, Er |
| 49 | 123 | Panama | Red Spanish | + | − | − | − | + | − | − | − | + | + | 1, A, Co, Er |
| 50 | 124 | Panama 1 | Taboga | + | − | − | − | + | + | − | − | − | + | 1, A, B, Er |
| 51 | 135 | Venezuela | Morada | + | − | − | − | + | − | − | − | + | + | 1, A, Co, Er |
| 52 | 136 | Venezuela 1 | Criolla | + | + | + | − | + | − | − | − | − | − | 1, 2, 3, A |
| 53 | 138 | Venezuela | Pina Lisa | − | − | − | − | − | + | − | − | + | + | B, Co, Er |
| 54 | 139 | Portugal 3 | Cayenne Azores | + | + | + | − | − | + | − | − | − | − | 1, 2, 3, B |
| 55 | 140 | Vietnam | Pakse | + | + | − | − | + | − | − | − | − | − | 1, 2, A |
| 56 | 142 | Vietnam 2 | Den | + | + | − | + | + | − | − | − | − | − | 1, 2, 6, A |
| 57 | 143 | Colombia | Pina De Castilla | + | + | − | − | + | − | − | − | + | − | 1, 2, A, Co |
| 58 | 145 | Puerto Rico 1 | Cabezona | − | + | − | − | + | − | − | − | − | − | 2, A |
| 59 | 149 | Brazil | CB 33 | − | − | + | − | − | − | − | − | − | + | 3 |
| 60 | 163 | Thailand | N91-05 | + | + | + | − | + | − | − | − | − | − | 1, 2, 3, A |
| 61 | 164 | Thailand | N91-06 | + | + | − | − | − | − | − | − | + | − | 1, 2, Co |
| 62 | 167 | USA: FL | 32419 | − | − | − | − | − | − | − | − | + | + | Co, Er |
| 63 | 173 | Bolivia | Short fruit #2 | − | + | + | − | + | − | − | − | + | + | 2, 3, A, Co, Er |
| 64 | 175 | Bolivia | Long fruit #2 | − | − | − | − | − | + | − | − | − | + | B, Er |
| 65 | 219 | USA: HI | + | + | + | − | − | − | − | − | − | − | 1, 2, 3 | |
| Total positives | 43 | 43 | 29 | 5 | 33 | 17 | 2 | 0 | 27 | 27 | ||||
| Prevalence (%) | 66.1 | 66.1 | 44.6 | 7.7 | 50.8 | 26.2 | 3.1 | 0.0 | 41.5 | 41.5 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larrea-Sarmiento, A.E.; Olmedo-Velarde, A.; Wang, X.; Borth, W.; Domingo, R.; Matsumoto, T.K.; Suzuki, J.Y.; Wall, M.M.; Melzer, M.J.; Hu, J. Genetic Diversity of Viral Populations Associated with Ananas Germplasm and Improvement of Virus Diagnostic Protocols. Pathogens 2022, 11, 1470. https://doi.org/10.3390/pathogens11121470
Larrea-Sarmiento AE, Olmedo-Velarde A, Wang X, Borth W, Domingo R, Matsumoto TK, Suzuki JY, Wall MM, Melzer MJ, Hu J. Genetic Diversity of Viral Populations Associated with Ananas Germplasm and Improvement of Virus Diagnostic Protocols. Pathogens. 2022; 11(12):1470. https://doi.org/10.3390/pathogens11121470
Chicago/Turabian StyleLarrea-Sarmiento, Adriana E., Alejandro Olmedo-Velarde, Xupeng Wang, Wayne Borth, Ryan Domingo, Tracie K. Matsumoto, Jon Y. Suzuki, Marisa M. Wall, Michael J. Melzer, and John Hu. 2022. "Genetic Diversity of Viral Populations Associated with Ananas Germplasm and Improvement of Virus Diagnostic Protocols" Pathogens 11, no. 12: 1470. https://doi.org/10.3390/pathogens11121470